
La enfermedad de Ollier o encondromatosis múltiple, descrita por el cirujano francés Louis Ollier (1830 – 1900), es una enfermedad ósea rara que afecta tanto a hombres como a mujeres.
Se desconoce la prevalencia exacta de esta enfermedad. Por extrapolación de un cierto número de pacientes conocidos en un departamento francés, se ha propuesto la cifra de 300 a 500 enfermos en Francia. En la actualidad, la única cifra oficial es el número de pacientes registrados por la asociación: 117. Desde 1998, cada año una decena de nuevas personas se ponen en contacto con la asociación.
La enfermedad es probablemente el resultado de una mutación somática (una mutación en un clon celular que se produce durante el desarrollo y no afecta a las células reproductoras). Sin embargo, no se ha identificado el gen o genes implicados. Se ha sugerido una cierta heterogeneidad genética (varias mutaciones posibles). A pesar de la mención de algunos casos familiares (tío-sobrina, hermanos, etc.), esta enfermedad no es hereditaria.
La enfermedad provoca lesiones óseas y se define por la coexistencia de al menos 3 condromas de localización asimétrica.
300
pacientes declarados en Francia. Cada año se registran 10 personas más.
Un condroma es una masa de tejido cartilaginoso en exceso que escapa a la regulación del crecimiento y perjudica el desarrollo normal del hueso.
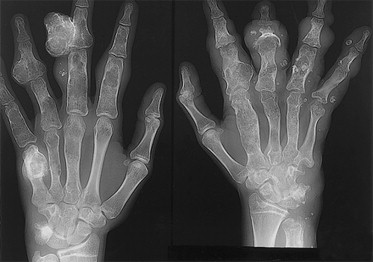
El rasgo característico de la enfermedad de Ollier es su extrema variabilidad clínica (tamaño, edad de aparición y diagnóstico de los condromas, número de condromas, curso, número de operaciones necesarias). La gravedad está relacionada con el número de condromas, la anchura del condroma en relación con la anchura de la metáfisis y la edad a la que aparecen los primeros condromas.
En la mayoría de los casos, los primeros signos de la enfermedad aparecen en la primera infancia, aunque se ha observado un diagnóstico tardío.
No todos los condromas implantados e identificados en una persona causan problemas sistemáticamente. Algunos permanecen inertes y no se desarrollan. Aún se desconocen los mecanismos que intervienen en la actividad o estabilidad de los condromas.
Los daños suelen predominar en un lado. En diversos grados y según las personas, pueden provocar deformidades, desviaciones axiales y diferencias en la longitud de los huesos.
No existe fragilidad ósea como tal (a diferencia de la osteoporosis, la osteogénesis imperfecta y la displasia fibrosa). Hay fragilidad local, debida a una pérdida de continuidad en la estructura ósea. Por tanto, si se produce un impacto y una fractura, ésta se producirá preferentemente en el condroma.
Algunos pacientes experimentan sensibilidad en determinados condromas (sobre todo en caso de impacto), mientras que otros experimentan un dolor más generalizado. Este dolor se debe a los condromas y a sus complicaciones, en particular a las compresiones que provocan (nervios) y a las deformaciones y desviaciones axiales resultantes. También se mencionan los dolores inherentes a los desequilibrios estaturales.
Cuando los angiomas se asocian a encondromatosis múltiples, se habla de síndrome de Maffucci (o síndrome de Kast o síndrome de Maffucci-Kast, descrito por el patólogo italiano Angelo Maffucci en 1881).
Un angioma es una lesión vascular localizada en el espesor de la piel o del tejido subcutáneo (grasa, músculo). En el caso de la enfermedad de Ollier-Maffucci, estas anomalías suelen ser más frecuentes en las manos; aparecen como cúmulos de carne vascularizados y violáceos.
Los angiomas también pueden causar dolor.
Tampoco se ha identificado la mutación o mutaciones implicadas en el síndrome de Maffucci.
Las pacientes han señalado problemas de fatiga. Esto puede estar relacionado en parte con la desventaja física asociada a los condromas (desviaciones, acortamiento), pero no debe pasarse por alto un componente psicológico (normal).
Se han observado tumores asociados en la enfermedad de Ollier y en el síndrome de Maffucci. La literatura médica cita varios casos de tumores benignos de ovario. Algunos miembros de la asociación se han visto afectados.